

Download a PDF of this statement
ASCRS CyPass Withdrawal Task Force
Leads: Douglas Rhee, MD; Nathan Radcliffe, MD; and Francis Mah, MD
Glaucoma: Leon Herndon, MD; Marlene Moster, MD; Thomas Samuelson, MD; Steven Vold, MD;
Ike Ahmed, MD
Cornea: Ken Beckman, MD, FACS; John Berdahl, MD; Marjan Farid, MD; Preeya Gupta, MD
Purpose
On August 29th, 2018, Alcon voluntarily withdrew the CyPass device from the market due to safety concerns reportedly based on 5-year data from the COMPASS XT study which indicate a higher rate of endothelial cell loss (ECL) in patients receiving cataract extraction (CE) plus CyPass versus CE alone. This ASCRS task force was convened to develop an understanding of the data and a preliminary consensus on monitoring and treatment options.
Overview of the Results Provided by Alcon
The results suggest a relationship between CyPass implant depth and ECL. Stents with greater anterior chamber exposure may have greater ECL at 5 years. Early migration of the implant has been observed; the potential for migration remains an important variable to be considered for long-term diagnostic monitoring.
The COMPASS XT study followed a smaller number of patients than the COMPASS trial. By 60 months, there were roughly 200 CyPass patients and 53 control patients. Of note, as the study was being assembled at 36 months, there are too few (n=36 patients) presenting at 36 months to make any meaningful comparisons. Aside from ECL, outlined below, there were no other significant safety concerns.
At 5 years, there was more ECL in CE with CyPass compared to CE alone (control). Baseline endothelial cell counts (ECCs) were 2432 for CyPass and 2434 for control, falling at 48 months to 1992 in CE with CyPass (n=116) vs 2303 in control (n=33) and at 60 months to 1931 in CE with CyPass (n=163) vs 2189 in the control group (n=40). This represented an 18.4% reduction in ECL in CE with CyPass vs 7.5% ECL in the control group at month 48, and to a 60-month CE with CyPass ECL of 20.5% compared to 10.1% in the control group (figure 1). The difference in ECL between CE with CyPass and control decreased slightly between 48 and 60 months. ANSI Z80:27 standards consider 30% ECL at 5 years to be meaningful. The percentage loss was 27.2% in CyPass vs 10% in controls (figure 2).
There appears to be a correlation between CyPass implantation depth and rate of ECL. In the COMPASS XT study, anterior chamber angle photos were taken, and the number of rings (figure 3) visible were used to grade implantation depth. There were 15 subjects in the COMPASS-XT trial where the number of visible rings was reported as changed between trial visits. Seven of these were reported to have more rings becoming visible with time, 7 were reported to have fewer visible rings with time, and there was 1 subject with an increase in visible rings followed by a decrease. It is possible that these changes were a result of observer variability.
For eyes with no rings showing (n=69), the rate was 1.39%/year, for 1 ring showing (n=98) 2.74%/year, and for 2-3 rings showing (n=27) 6.96%/year (figure 4). No patients in COMPASS XT required corneal surgery by 5 years. Four patients underwent a CyPass trimming procedure for a CyPass with 3 rings visible in the anterior chamber that was observed in the first postoperative week. In all cases, the corneas remained clear and the ECC remained stable at month 60. One patient in the COMPASS trial (two-year follow up) did undergo a Descemet’s stripping endothelial keratoplasty (DSEK) at month 13 with the procedure being thought to be related to the CE and not to the CyPass, which was well positioned with 1 ring visible. Some eyes with >2 rings visible in the anterior chamber experienced minimal ECL (figure 5). Thus, clinically relevant, judicious, and periodic monitoring of corneal health is advised.
Summary of Task Force Discussion of Results: The ECL in the control group in the COMPASS XT study experienced a very low rate of ECL, even compared to historic norms. The previously reported pseudophakic ECL rate is roughly 2%/year; a rate of 0.36%/year ECL was observed in control Compass XT patients. Additionally, it was noted that, depending on glaucoma severity, cataract alone may not be the best comparison group for eyes receiving CE with CyPass device, but rather eyes receiving CE and other glaucoma drainage procedures.
Diagnostic Monitoring Options
Notification:
Physicians who have implanted CyPass devices should refer to their hospitals, ASC’s, or practice policies regarding patient notification for medical implants that have been voluntarily withdrawn from the market. Patients who have received CyPass devices should continue to be followed by their eye care provider(s) at appropriate intervals.
Risk Assessment and Diagnostic Monitoring:
Gonioscopy should be performed on patients with an indwelling CyPass device with attention to device position - the presence or absence of contact between the corneal endothelium and the CyPass device, the position of the device lumen anterior to Schwalbe’s line, and to the number of retention rings visible in the anterior chamber.
The group noted that numerous conditions and therapeutic interventions can result in ECL, but intervention is generally limited to when clinically apparent or functionally significant changes occur. Slit lamp examination to assess for focal or diffuse corneal stromal edema and/or presence of guttata is recommended. Symptoms of morning blurriness or increasing glare with bright lights could be indicative of clinically/functionally significant corneal edema, but there are numerous confounding conditions with these same symptoms limiting the diagnostic value of symptomatology alone.
If quantification is desired, baseline and follow-up corneal pachymetry, for corneal thickness measurements, as well as specular microscopy, for endothelial cell counts, could be considered. However, both of these methodologies have significant variability of measurement that are inherent to the techniques--e.g., variations in measurements can occur due to lack of consistency in applanation location among each measurement taken. Clinical examination alone may be appropriate for monitoring patients with indwelling CyPass devices.
When to Consider Intervention
The CyPass micro-stent Instructions For Use (IFU) states, “in the absence of clinical sequelae, device adjustment or removal is not recommended.” (Ref: https://www.accessdata.fda.gov/cdrh_docs/pdf15/p150037d.pdf) According to the CyPass IFU, “Situations that may merit consideration of CyPass Micro-Stent position adjustment or removal include, but are not limited to: intermittent or persistent contact between the CyPass Micro-Stent and the corneal endothelium; significant decrease in endothelial cell density that appears related to CyPass Micro-Stent positioning or stability; iris-cornea touch; persistent hypotony; persistent uncontrolled uveitis; recurrent or persistent hyphema with IOP elevation above target pressure; or any anatomic or functional clinical sequelae of the anterior or posterior segment that may cause a threat to vision.”
Eyes with 0 or 1 ring of the CyPass device visible in the anterior chamber by gonioscopy: Without clear evidence of corneal decompensation, the consensus was that no action other than clinical monitoring is recommended.
Eyes with 2 or 3 rings of the CyPass device visible in the anterior chamber by gonioscopy:
In these eyes, there is a greater risk of corneal ECL. However not all eyes in this category will experience clinically meaningful ECL. Without clinically significant evidence of corneal decompensation, no action other than monitoring is indicated. More frequent corneal evaluation could be considered.
Considerations for Device Revision
If corneal decompensation develops and >1 ring of the device is visible, the surgeon may consider CyPass repositioning, removal, or proximal end trimming. It was the consensus of the group that implant repositioning i.e. deeper implantation, would be most safe if performed within 7-10 days of implantation. Beyond this time period, there was concern expressed by the group that fibrosis around and/or through the filtration holes of the device may create a higher risk of complications with device repositioning. Due to the potential for fibrosis around and possibly investing the device, device removal was not favored by the group. Trimming of the proximal end is likely to be the preferred procedure if the patient and physician desire intervention. Technical descriptions of the procedures are described in the CyPass micro-stent Instructions for Use “IFU.”
Figures: These figures are public information and found on Alcon’s website.

Figure 1: Percent Change in Endothelial Cell Density from Baseline, by Visit and Treatment Group Error bars indicate 95% confidence intervals
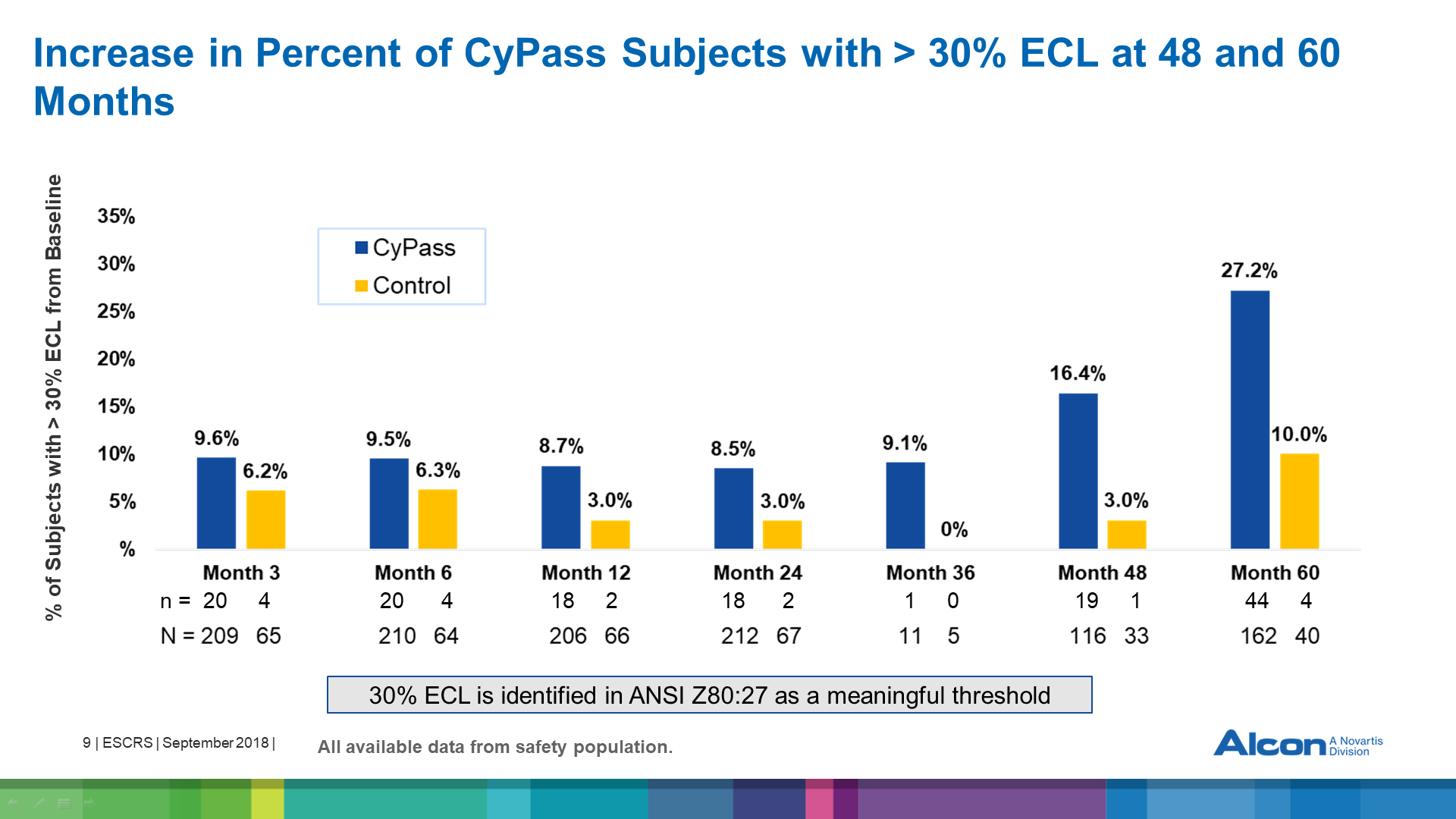
Figure 2: Percentage of eyes that experienced a >30% reduction of endothelial cell count as measured by specular microscopy

Figure 3: Schematic Image of the CyPass Device. The line denoting the 3 retention rings points to the middle of the three rings. Image source (Your CYPASS Ultra System Quick Reference Guide; Alcon, Ft. Worth TX)
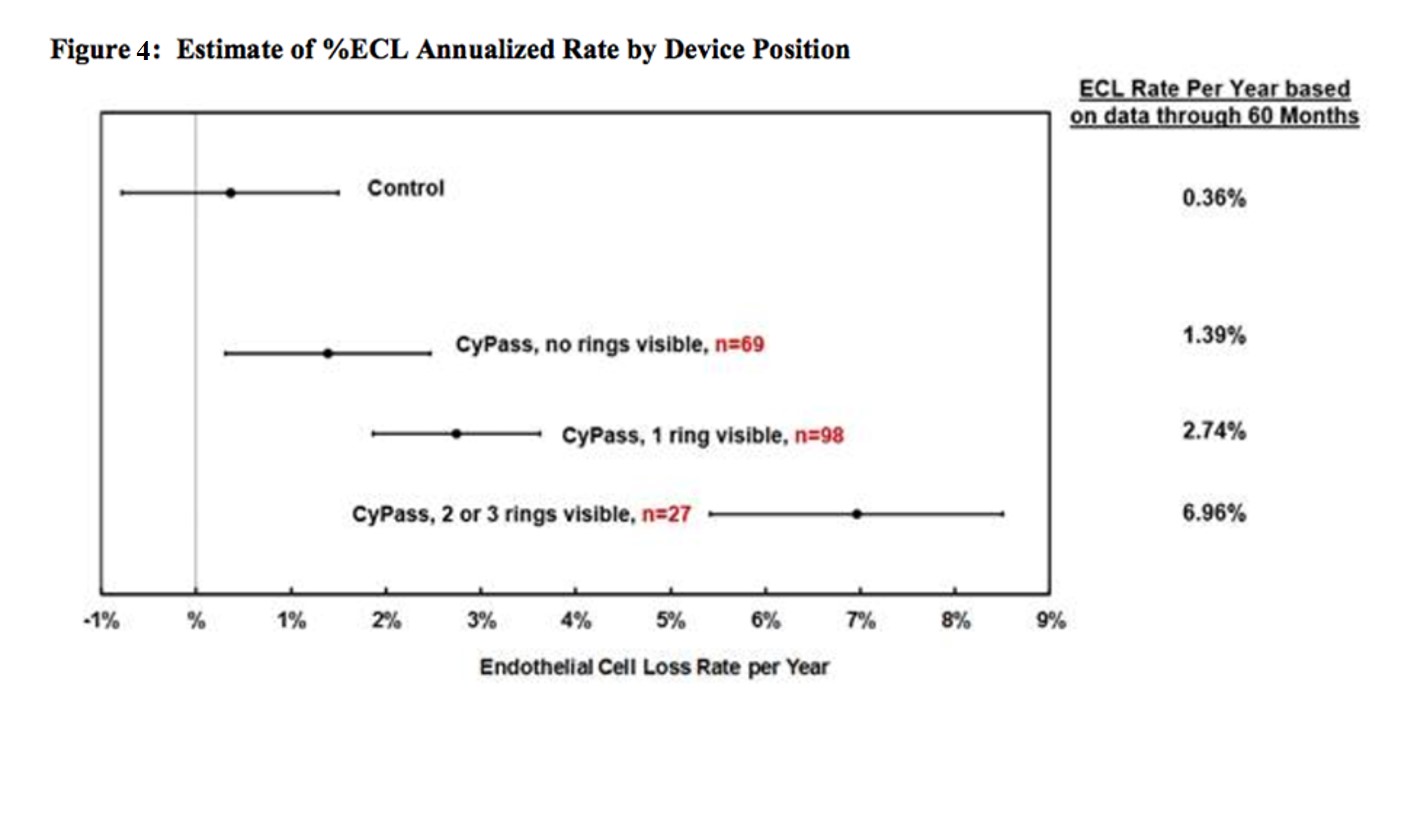
Figure 4: Estimate of %ECL Annualized Rate by Device Position.” The mean and standard deviation of percentage loss of endothelial cells as measured by specular microscopy are represented based on the amount of the CyPass device that was visible in the anterior chamber by gonioscopy.
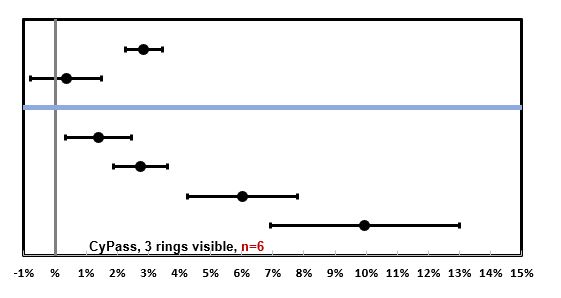
Figure 5: Among the eyes with 3 of the retention rings of the CyPass device visible in the anterior chamber, there was significant variability in the rate of endothelial cell loss (ECL).
A more detailed overview of the results from the 5-year follow-up study of the CyPass Micro-stent is available at www.alcon.com/CyPass.
DISCLAIMER: This consensus statement is provided by ASCRS for informational and educational purposes only and is intended to offer practitioners recommended monitoring and treatment options for cataract patients who received the CyPass implant. Practitioners should use their personal and professional judgment in interpreting these recommendations and applying them to the particular circumstances of each affected cataract patient. This document is not intended to provide medical advice, create a standard of care, or be deemed inclusive of all proper methods of care nor exclusive of other methods of care reasonably directed to obtaining the same results. Adherence to these recommendations will not ensure successful treatment in every situation. The information in this statement is provided “as is,” and ASCRS makes no warranties as to its accuracy or completeness. This consensus statement may need to be updated as future studies on the effect of the CyPass implant are conducted.